Jump to: Current Lab Projects
HIV and cardiovascular diseases
It has long been established that individuals infected with Human immunodeficiency virus type 1 (HIV-1) are at an increased risk of developing atherosclerosis and cardiovascular disease. Compared with uninfected individuals, people suffering from HIV infection have a 1.5- to twofold higher incidence of cardiovascular events reported. This observation was originally attributed to antiretroviral therapy, however, increasing evidence suggests that HIV infection itself contributes significantly to the risk of coronary artery disease (CAD). Chronically elevated cholesterol is considered to be the driving force behind atherosclerosis and CAD, but chronic inflammation is also an essential element of the pathogenesis of atherosclerosis. It has recently been demonstrated that HIV infection interferes with the cholesterol metabolism of the human host cells, in particular macrophages, thereby elevating the risk of atherosclerosis. However, the mechanisms of this connection are not completely understood.
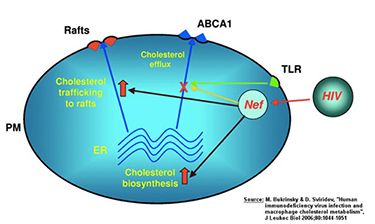
Many viruses, including HIV, require cholesterol for their replication and as a structural element. One mechanism of how HIV infection may accelerate the development of atherosclerosis is by inhibiting the emission of cellular cholesterol. To alter the cellular cholesterol content, the virus “hijacks” the pathways responsible for maintaining intracellular cholesterol metabolism. Via its accessory protein Nef, HIV inhibits the activity of the lipid transporter ABCA1, a key element of the cholesterol discharge machinery. This way HIV promotes accumulation of cholesterol in macrophages and their transformation into foam cells, which are a hallmark of atherosclerosis. Not only does Nef impair cholesterol efflux in macrophages infected by HIV, but soluble Nef, which is released from infected cells, also has a systemic effect impairing ABCA1 in cells not infected with the virus. How this capacity of Nef relates to cellular pathways involved in maintaining cholesterol homeostasis and, specifically, why and how Nef inhibits ABCA1 remains unclear. The involvement of these pathways may not be limited to HIV infection, as they may represent a physiological regulation of cellular cholesterol metabolism exploited by HIV and possibly by other infections.
The HIV protein Vpr and its interactions with the host cell
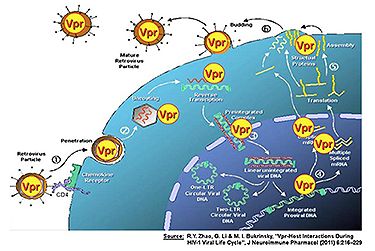
HIV-1 is engaged in dynamic and antagonistic interactions with the human host cells. Once infected by HIV-1, host cells initiate various antiviral strategies to counteract the viral invasion. In its turn, the virus has different strategies to suppress these host responses to infection. The final balance between these interactions determines the outcome of the viral infection and disease progression.
The HIV accessory protein viral protein R (Vpr) is a multifunctional protein that plays an important role at multiple stages of the HIV-1 viral life cycle. Vpr is required for efficient viral replication in non-dividing cells such as macrophages, and it promotes viral replication to some extent in proliferating CD4+ T cells. Although the molecular mechanisms underlying these activities are subject of ongoing investigations, these activities have been linked to the promotion of viral replication and impairment of anti-HIV immunity.
Recent findings suggest that Vpr interacts with some of the host innate antiviral factors, such as heat shock proteins, and plays an active role as a viral pathogenic factor. Cellular heat stress response factors counteract Vpr activities and inhibit HIV replication, however, Vpr overcomes these heat-stress-like responses. Importantly, functional defects of Vpr have been correlated with slow disease progression of HIV-infected patients. Vpr plays a pivotal role in viral pathogenesis since Vpr activities are linked to the promotion of viral infection. Strategies to inhibit these adverse Vpr effects could potentially alleviate the impact of the virus and benefit infected patients. Therefore, finding cellular factors that are capable of restricting Vpr activities could provide an opportunity for possible alternative anti-HIV therapy.
The role of cyclophilins in inflammatory diseases
CD147 is a ubiquitously expressed integral plasma membrane glycoprotein, which has been shown to regulate multiple physiological functions, including lymphocyte responsiveness, monocarboxylate transporter expression and spermatogenesis. Recently, CD147 was also demonstrated to function as a signaling receptor for the extracellular cyclophilins A and B, and to mediate chemotactic activity of cyclophilins towards a variety of immune cells.
Studies using multiple model systems have demonstrated a role for cyclophilin–CD147 interactions in the regulation of inflammatory responses in a number of human diseases, including acute lung inflammation, rheumatoid arthritis, and cardiovascular disease. Given the role that cyclophilin–CD147 interactions play in pathogenesis of inflammatory diseases, they present an attractive target for therapeutic interventions. Therefore, it will be extremely important to refine our understanding of cyclophilin-induced signaling through CD147.
Agents targeting either CD147 or cyclophilin activity show significant anti-inflammatory effects in experimental models, suggesting CD147–cyclophilin interactions may be a good target for new anti-inflammatory therapeutics. The current challenge is to design therapeutic agents with the capacity to block specific functions of these molecules while leaving other functions unaffected.
Current Lab Projects
Studies in the Bukrinsky laboratory focus on two medically important areas: HIV infection and inflammation. In HIV research, we focus on so-called accessory proteins of HIV, Vpr and Nef, which are responsible for many pathologic effects of infection. For the Nef protein, we investigate the innovative idea that connects this protein to the high risk of cardio-vascular disease observed in HIV-infected patients. Our studies are designed to identify the molecular mechanisms behind the discovered in our laboratory inhibitory effect of Nef on cellular cholesterol transporter ABCA1. These studies are expected to identify the missing link between HIV infection and atherosclerosis and will provide rationale for developing novel therapeutic agents that can be used to treat atherosclerosis in HIV patients. Moreover, understanding the effects of HIV infection on cholesterol homeostasis will provide new approaches to anti-HIV therapy, prevention, and cure.
Another line of HIV-related research in the Bukrinsky laboratory is focused on identification of a host cell anti-HIV restriction factor targeted by HIV-1 accessory protein Vpr. Most HIV accessory proteins (there are 5 such proteins in HIV-1 and HIV-2) have evolved to target anti-viral restriction factors, interferon-inducible proteins that protect us from viral infections. Vpr remains the only accessory protein without a specific target within the host cell. We have developed an innovative method that allows the detection of this target. Identification of this new restriction factor will not only advance our understanding of anti-HIV immunity but will provide novel targets for anti-HIV therapy and vaccine design.
Finally, our work on inflammation focuses on the new inflammatory factor that we discovered – extracellular cyclophilins. We continue studying the role of extracellular cyclophilins in different inflammatory diseases (e.g. atherosclerosis, rheumatoid arthritis), with specific emphasis on novel therapeutic agent that we developed – a cyclosporine A analog that does not enter cells. This innovative drug specifically targets extracellular cyclophilins, is not immunosuppressive, and does not have side effects characteristic to cyclosporine A while showing promising results in reducing inflammation. We expect this drug to become a treatment of choice in many inflammatory diseases.